Raman Spectroscopy, Group Theory, and Computational Chemistry: A Physical Chemistry Laboratory Experiment on para-Difluorobenzene
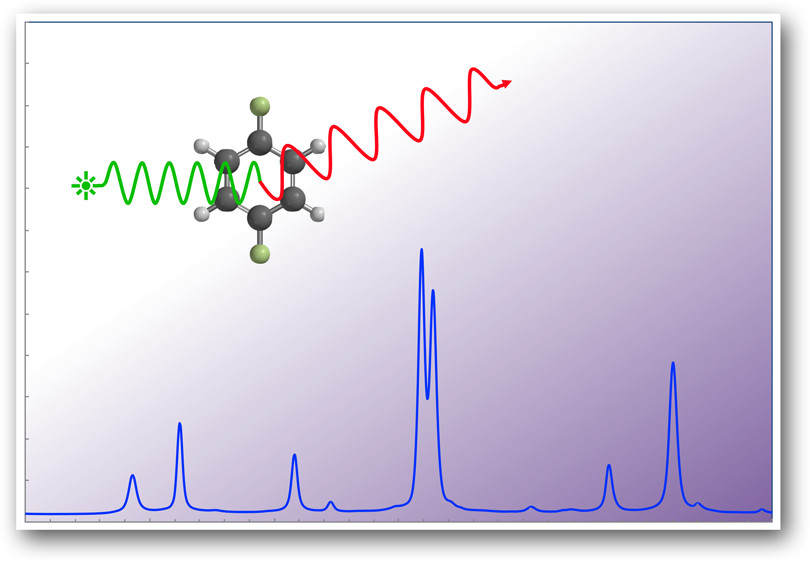
An experiment for the undergraduate physical chemistry laboratory is described in which the Raman spectrum of liquid para-difluorobenzene is recorded and assigned. A density functional theory (DFT) calculation of the 30 normal modes of the molecule is undertaken using computational chemistry software. Students use group theory to determine which of the modes are Raman active. Guided by these results, they are able to identify 13 of the 15 symmetry-allowed fundamental bands in their Raman spectrum. The remaining 17 observed bands are assigned as either overtone or combination bands, by estimating the wavenumbers of symmetry-allowed transitions from the scaled results of their DFT calculation. The experiment demonstrates the powerful way in which group theory and computational chemistry can be used to assign a dense Raman spectrum of an organic compound.
The acquisition and measurement of the Raman lines of pDFB takes about 45 min, whereas the data analysis requires 3–4 h, depending on how fast a team works. Students begin their vibrational analysis by identifying the symmetries of the vibrational normal modes. They then complete a density functional theory (DFT) calculation of the Raman spectrum using the hybrid functional B3LYP with a 6-311G* basis set. With Spartan 20 software on a laptop computer, this calculation typically takes less than 10 min.
Citation
Thomas D. Varberg, Journal of Chemical Education, 2022, 99, 5, 2129-2134, https://doi.org/10.1021/acs.jchemed.2c00095
License
Copyright 2022 American Chemical Society and Division of Chemical Education, Inc.