Best Practice DFT Protocols for Basic Molecular Computational Chemistry
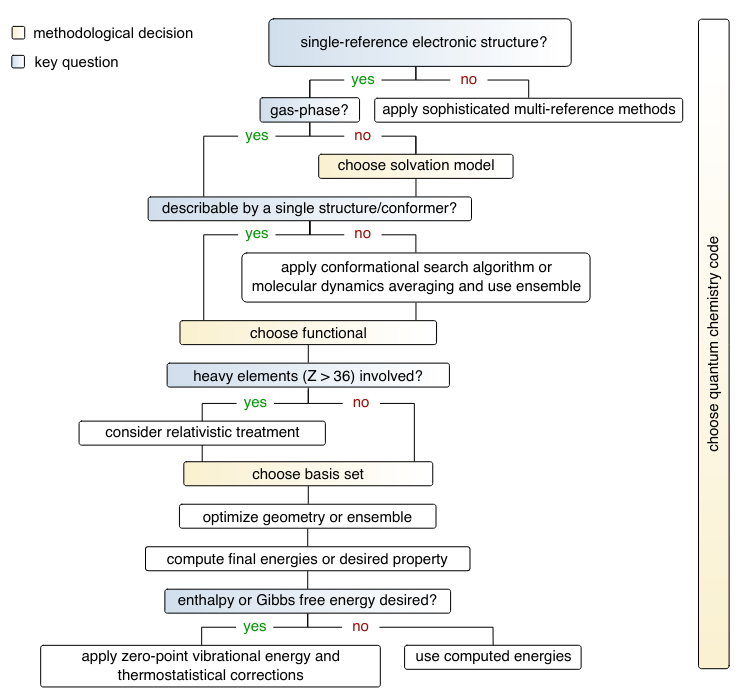
Nowadays, many chemical investigations can be supported theoretically by routine molecular structure calculations, conformer ensembles, reaction energies, barrier heights, and predicted spectroscopic properties. Such standard computational chemistry applications are most often conducted with density functional theory (DFT) and atom-centered atomic orbital basis sets implemented in many standard quantum chemistry software packages. This work aims to provide general guidance on the various technical and methodological aspects of DFT calculations for molecular systems, and how to achieve an optimal balance between accuracy, robustness, and computational efficiency through multi-level approaches. The main points discussed are the density functional, the atomic orbital basis sets, and the computational protocol to describe and predict experimental behavior properly. This is done in three main parts: Firstly, in the form of a step-by-step decision tree to guide the overall computational approach depending on the problem; secondly, using a recommendation matrix that addresses the most critical aspects regarding the functional and basis set depending on the computational task at hand (structure optimization, reaction energy calculations, etc.); and thirdly, by applying all steps to some representative examples to illustrate the recommended protocols and effect of methodological choices.
Citation
Bursch M, Mewes J-M, Hansen A, Grimme S. Best Practice DFT Protocols for Basic Molecular Computational Chemistry. ChemRxiv. Cambridge: Cambridge Open Engage; 2022; This content is a preprint and has not been peer-reviewed.
https://doi.org/10.26434/chemrxiv-2022-n304h
License
The content is available under CC BY NC ND 4.0 License Creative Commons